
Fucoidan, a type of polysaccharide, is obtained through extraction from various kinds of marine brown algae. Fucoidan has been documented to exhibit antitumor, antioxidant, antiviral, antibacterial, anti-inflammatory, and anticoagulant effects. Fucoidan is recognized for its ability to trigger significant apoptosis in a range of cancer cell lines; however, the precise molecular pathways responsible for this effect remain incompletely understood.
This blog post will discuss the study “Fucoidan Extract Induces Apoptosis in MCF-7 Cells via a Mechanism Involving the ROS-Dependent JNK Activation and Mitochondria-Mediated Pathways” by Zhongyuan Zhang et al. The research indicates that fucoidan effectively suppresses the growth of MCF-7, MDA-MB-231, HeLa, and HT1080 cells, yet exhibits minimal toxicity to the MCF-10A human mammary epithelial cell line.
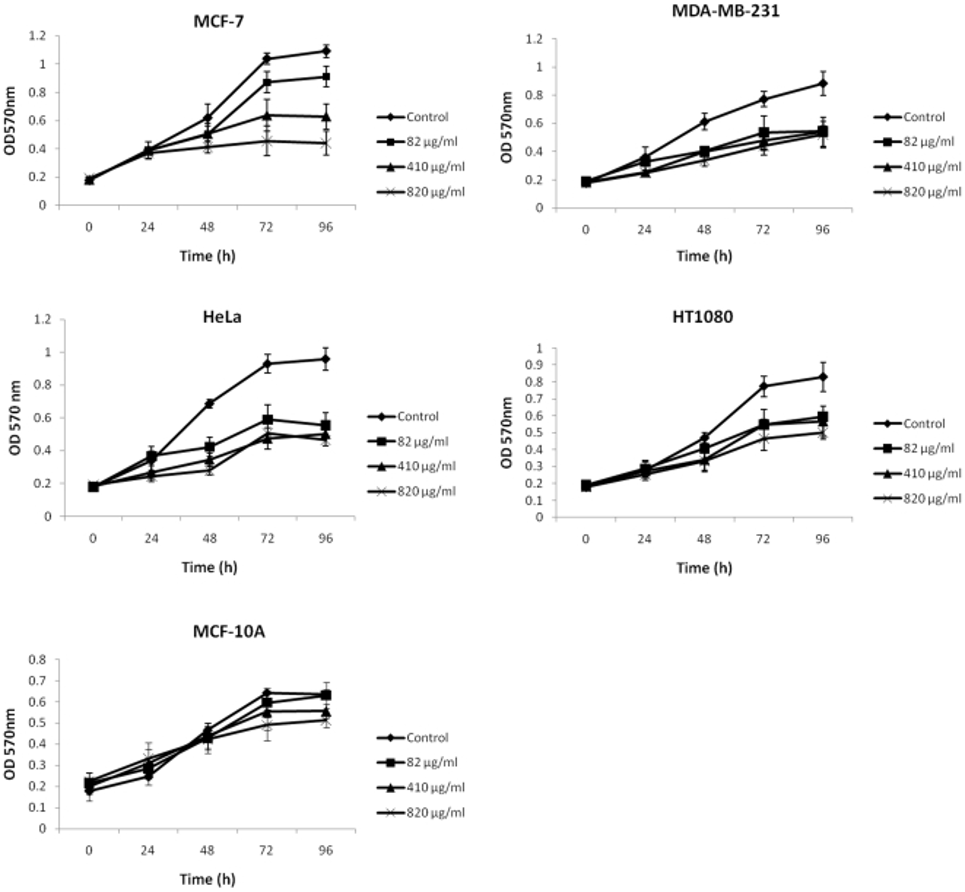
Researchers employed an MTT assay to determine the effects of low-molecular-weight fucoidan (FE) on numerous cancer cell lines. Cells were subjected to various concentrations of FE for a duration of 96 hours. As shown in Figure 1, FE treatment inhibited cell proliferation in MCF-7 cells (60% growth inhibition), MDA-MB-231 cells (41% growth inhibition), HeLa cells (52% growth inhibition), and HT1080 cells (40% growth inhibition). MCF-7 cells were significantly more sensitive to different doses of FE than the other three cell lines. The nonmalignant MCF-10A cell line was less sensitive to FE treatment than the malignant cell lines. The evidence indicates that FE plays a role in significantly hindering the growth of human cancer cells.
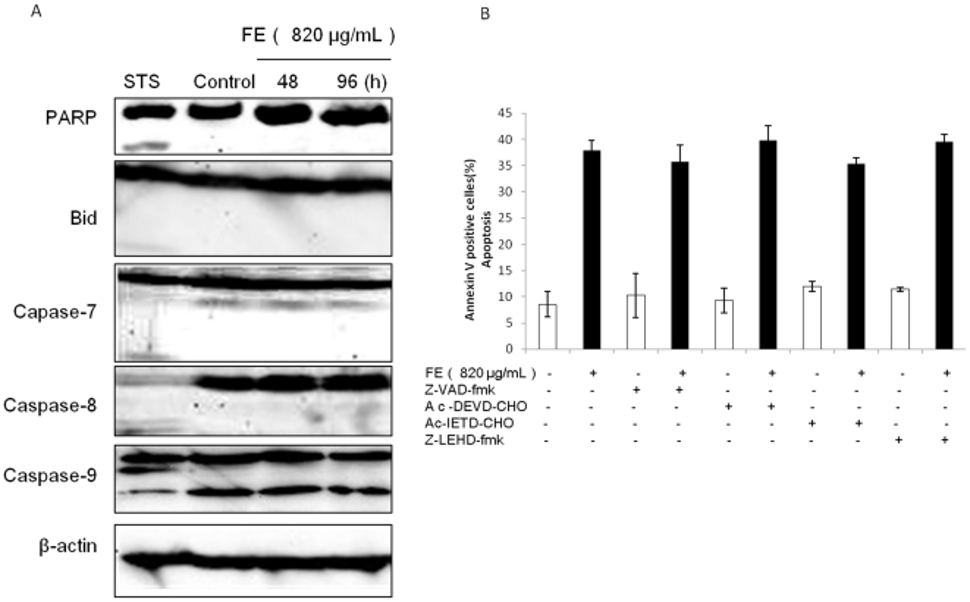
Despite the established lack of caspase-3 expression in MCF-7 cells, the study utilized Western blotting to analyze other caspase family members and determine their potential role in FE-mediated apoptosis. In the experiments, we found no significant increase in caspase-9 cleavage, nor activation of PARP, Bid, caspase-7, or caspase-8 in FE-treated MCF-7 cells (Figure 2A). Furthermore, we performed caspase inhibition assays using the general caspase inhibitor Z-VAD-fmk and specific inhibitors for caspase-3/7, caspase-8, and caspase-9, as determined by Annexin V/PI double staining analysis. However, as shown in Figure 2B, none of the caspase inhibitors attenuated FE-induced apoptotic cell death. Furthermore, Z-VAD-fmk did not have a protective effect against FE-induced phosphatidylserine exposure. These results suggest that caspase activation is not required for FE-induced apoptosis in MCF-7 cells.
The mitochondrial membrane potential (MTP) is crucial for controlling the permeability of the mitochondrial membranes, and it also significantly contributes to the initiation of apoptosis. Therefore, they examined the effect of FE on ΔΨm using rhodamine 123 (Rh123) stain. FE treatment of MCF-7 cells induced a loss of ΔΨm. Time-course analysis revealed that ΔΨm was lost in a significant number of FE-treated MCF-7 cells. Bcl-2 family proteins have been reported to regulate MMP. As a result, they studied the time-varying expression of Bcl-2 family proteins within MCF-7 cells exposed to FE. Western blotting analysis indicated that FE treatment reduced the levels of anti-apoptotic proteins like Bcl-2 and Bcl-xl, and somewhat elevated the levels of pro-apoptotic proteins such as Bax and Bad.
Furthermore, the ratio of Bax to Bcl-2 was measured by quantification of the bands. FE treatment caused a time-dependent increase in the Bax/Bcl-2 ratio in MCF-7 cells. Next, we established control (MCF-7/Vec) and Bcl-2-overexpressing MCF-7 cell lines (MCF-7/Bcl-2). MCF-7/Bcl-2 cells expressed significantly higher levels of Bcl-2 than control cells. Furthermore, FE-treated MCF-7/Bcl-2 cells consistently exhibited lower apoptosis rates compared with similarly treated MCF-7/Vec cells. Based on this data, FE treatment resulted in mitochondrial dysfunction within MCF-7 cells.
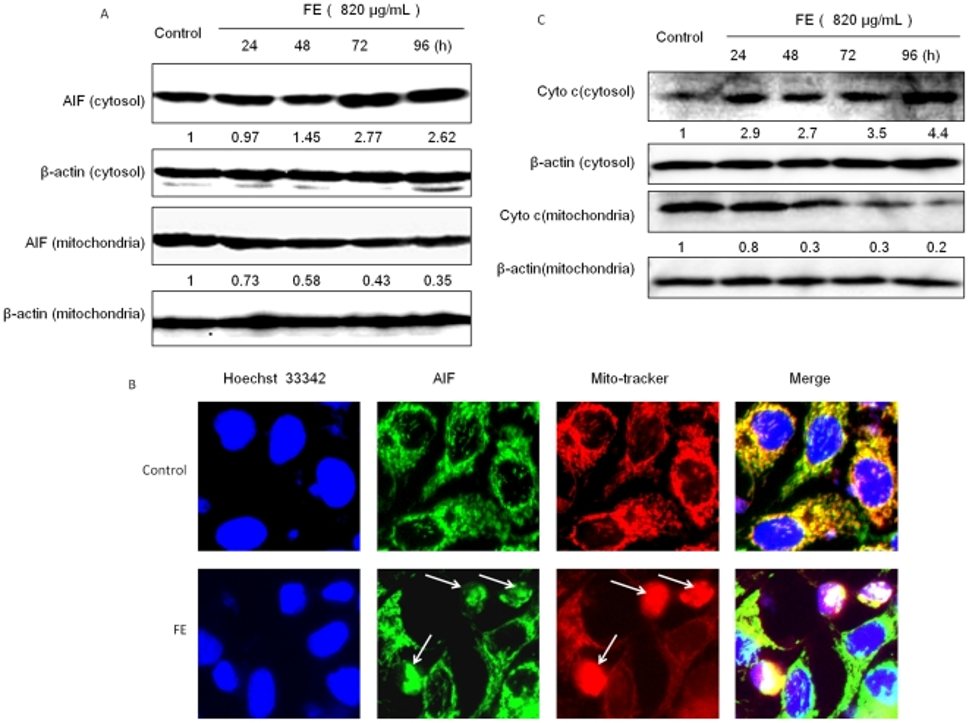
The release of mitochondrial apoptotic factors into the cytoplasm is a common consequence of drug-induced apoptosis that results in mitochondrial damage. Therefore, they investigated whether AIF plays a role in FE-induced apoptosis. Figure 3A illustrates that Western blotting revealed FE treatment promoted AIF’s translocation from mitochondria to the cytoplasm, with this effect intensifying over time. AIF localization was determined by immunofluorescence using specific antibodies. Mitochondria and nuclei were labeled with MitoTracker and Hoechst 33342, respectively. As shown in Figure 3B, untreated MCF-7 cells showed AIF staining in a punctate pattern, indicating normal mitochondrial localization and normal nuclear morphology. In contrast, FE-treated MCF-7 cells showed increased diffuse AIF staining in the nucleus, chromatin condensation, and nuclear fragmentation, characteristics typically associated with apoptosis. Furthermore, Western blotting detected cytochrome c, another apoptosis-regulating protein. The time-course analysis revealed that cytochrome c was released from the mitochondria into the cytoplasm, as depicted in Figure 3C.
The MAPK family is involved in the process of inducing cell death. Therefore, they evaluated the effect of FE on the phosphorylation of JNK, p38, and ERK1/2. MCF-7 cells were first treated with FE at a concentration of 820 µg/mL for various periods of time, and the phosphorylation of JNK, p38, and ERK1/2 was measured by Western blot analysis. Phosphorylation of JNK, p38, and ERK1/2 was detected as early as 30 minutes after FE treatment and persisted for at least 6 hours. FE’s presence led to the activation of the JNK, p38, and ERK1/2 signaling pathways in MCF-7 cancer cells, as established by these results. To assess whether these FE-induced changes are related to apoptosis, we examined the effects of specific inhibitors on MCF-7 cancer cells in the following study. Across the various inhibitors tested, the number of apoptotic cells was significantly reduced in cells treated with FE for 48 hours in the presence of the JNK-selective inhibitor SP600125. However, pretreatment with a p38 inhibitor (SB203580) or an ERK inhibitor (PD98059) had little effect. Pretreatment with SP600125 slightly reduced JNK phosphorylation 6 hours after FE treatment.
The study involved assessing intracellular ROS production in MCF-7 cells treated with FE, utilizing the DCFH-DA fluorescent probe for measurement. When cells were treated with FE, DCFH-DA-derived fluorescence increased 1.5-fold after the first 30 minutes, then steadily decreased, reaching below control levels within 6 hours. Furthermore, pretreatment with the scavenger NAC almost completely inhibited ROS production in a time-dependent manner. To clarify whether ROS are involved in apoptosis, we tested the effect of NAC on FE-treated MCF-7 cells and found that pretreatment with NAC partially inhibited the accumulation of apoptotic cells after 48 hours of FE treatment compared with cells treated with FE alone.
Subsequently, a thorough examination of when reactive oxygen species (ROS) were produced revealed that fluorescence started to rise after 15 minutes, reaching its highest point at 30 minutes. Phosphorylation of JNK, p38, and ERK1/2 was detected at 30 minutes and persisted for at least 6 hours. They compared this time course with the changes in ΔΨm induced by FE in MCF-7 cells. Considering these results, we investigated whether ROS generation occurs concomitantly with the phosphorylation of JNK, p38, and ERK1/2 and the decrease in ΔΨm in FE-treated MCF-7 cancer cells. Pretreatment with 2 mM NAC partially attenuated the phosphorylation of JNK, p38, and ERK1/2 6 hours after FE treatment. This suggests that ROS are required for the phosphorylation of JNK, p38, and ERK1/2. Furthermore, pretreatment with either SP600125 or NAC partially inhibited the disappearance of ΔΨm 48 hours after FE treatment. The sequence of events suggests that in FE-treated MCF-7 cells, ROS-dependent JNK phosphorylation occurs before mitochondrial membrane permeabilization.
The researchers investigated the regulatory role of FE in cellular metabolism by measuring intracellular ATP levels in MCF-7 cells. Six hours after FE treatment, a decrease in intracellular ATP levels was observed in MCF-7 cells. When FE was administered in a glucose-free medium, the decrease in ATP levels was even more pronounced. Annexin V-FITC staining, on the other hand, revealed that the induction of cell death occurred later than the decrease in ATP levels. Twenty-four hours after FE treatment, a clear increase in annexin V-FITC staining was observed, regardless of the rate of ATP depletion.
Based on these results, FE appears to activate a caspase-independent apoptotic process in MCF-7 cancer cells, driven by ROS-dependent MAP kinase activation and influence over the mitochondrial pathway by way of Bcl-2 family proteins.
Source: PLoS One. 2011 Nov 11;6(11):e27441. doi: 10.1371/journal.pone.0027441