
In this blog, I would like to inform you of the study, “A Fucus vesiculosus(FVE)extract inhibits estrogen receptor activation and induces cell death in female cancer cell lines” by Jianqing Zhang et al, which aimed to evaluate the potentially biologically relevant antitumor effects of the brown alga Fucus vesiculosus in estrogen receptor (ER)-dependent and -independent female cancer cell lines.
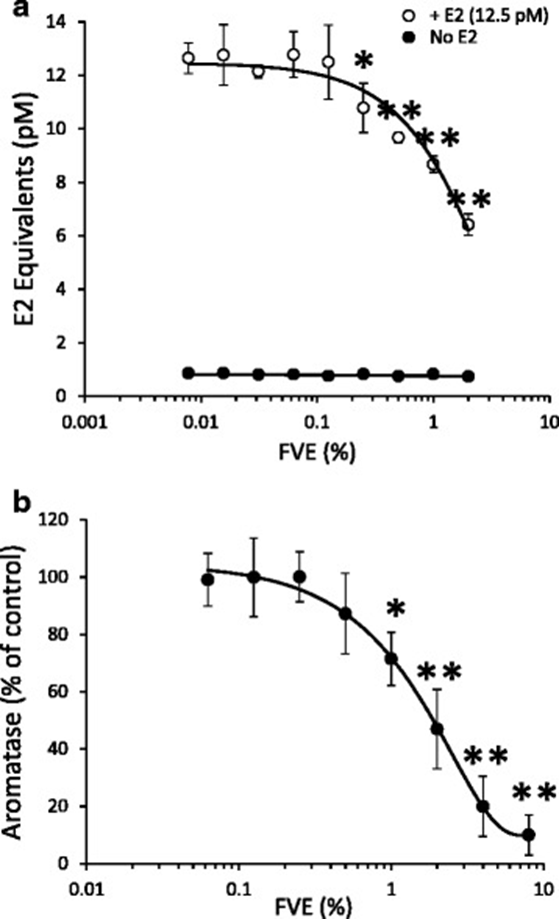
First, using the Calux assay, they found that FVE significantly reduced luciferase reporter activation by up to 50% in cells co-treated with 12.5 pM (EC50) E2 (Figure 1a). This suggests that FVE is a potent antagonist of E2-induced ER activation. Furthermore, in vitro enzymatic activity of aromatase was inhibited by FVE with an IC50 of 2% (Figure 1b). However, fucoidan did not alter ER activation or aromatase activity, even at concentrations exceeding the 10% dry weight reported for F. vesiculosus. The dual effects of antagonizing ER activation by circulating E2 and reducing E2 synthesis through aromatase inhibition suggest that F. vesiculosus may play a protective role in the development and progression of estrogen-dependent cancers.
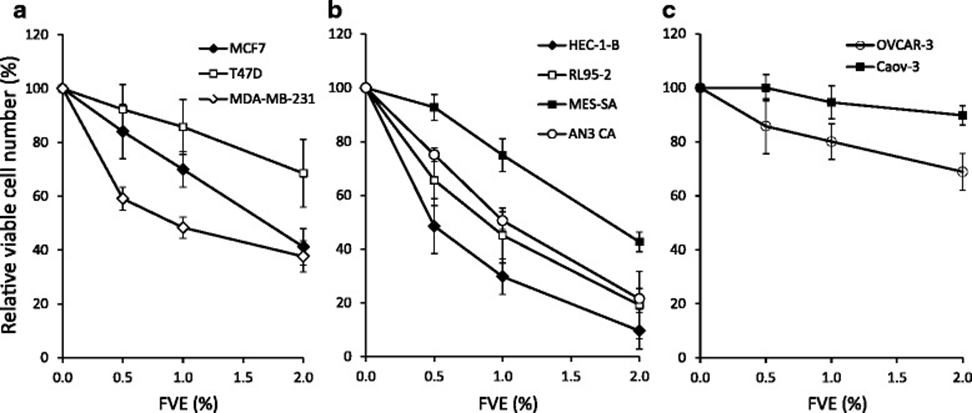
FVE treatment dose-dependently reduced cell viability after 3 days in most cancer cell lines tested, as measured by the MTT assay for mitochondrial activity. However, sensitivity varied significantly among cell lines (Figure 2a–c). FVE reduced cell viability most significantly in HEC-1-B and RL95-2 endometrial cells (Figure 2b). In contrast, no significant effect on cell viability was observed in Caov-3 ovarian cancer cells (Figure 2c). The cell line-specific sensitivity suggests that FVE is not toxic over the concentration range but rather induces cell death via a regulated pathway. FVE toxicity was further investigated using dual assays of mitochondrial ATP production and plasma membrane permeability. Treatment with FVE at the same concentrations (up to 2%) that have been shown to reduce cell viability in MCF-7 breast cancer cells did not produce toxic effects, compared with the impact of positive markers for mitochondrial toxicity (CCCP) and membrane permeability (digitonin), indicating the absence of a necrotic effect due to primary toxicity. During the cell proliferation experiment, morphological changes were observed within the cells after 72 hours of treatment with 1% FVE, primarily large cytoplasmic vesicles.
Gene expression profiling studies revealed that FVE-treated breast, endometrial, and ovarian cells, compared with untreated controls, exhibited differentially altered transcript levels, primarily in genes involved in apoptosis (APAF1, CASP6, FANCG, MED1, XIAP), autophagy (ATG10, GABARAP), and protein kinase-regulated signaling pathways (BRAF, MAP3K14, PIK3R4, PRKAA1, PRKACB, PRKAR1A, PRKAR2A). These results suggest that the FVE-induced decrease in cell viability is due to the induction of apoptosis and/or autophagy, and the morphological observations of cancer cells exposed to FVE also support the initiation of apoptotic and/or autophagy-mediated cell death.
They investigated the contribution of apoptosis and autophagy to the loss of viability of cancer cells exposed to FVE in MCF-7, MDA-MB-231, HEC-1-B, MES-SA, AN3-CA, OVCAR-3, and Caov-3 cells by combined treatment with the pan-caspase inhibitor VAD and the autophagy inhibitor 3-MA. Combined treatment with VAD reversed the effect of FVE on cell viability in MDA-MB-231, AN3-CA, and OVCAR cells. To further address the contribution of apoptosis to the loss of cell viability induced by FVE, we measured caspase activity in MCF-7 and MDA-MB-231 cells, which represent VAD-nonresponsive and VAD-responsive cell lines, respectively. FVE did not affect caspase activity in MCF-7 cells. However, FVE-treated MDA-MB-231 cells increased caspase activity, which was reduced by the addition of VAD. PARP cleavage by caspase 3 is a hallmark of apoptosis and caspase activation. Western blotting using an antibody specific for cleaved PARP revealed no significant PARP cleavage in MCF-7 cells, but a concentration-dependent increase in PARP cleavage was observed in MDA-MB-231 cells treated with FVE. These studies suggest that apoptosis is not the primary mechanism underlying the cell-killing effect of FVE on MCF-7 cells, but that apoptosis plays a role in cell death in MDA-MB-231 cells.
Co-treatment of cells with FVE and 3-MA reduced the effect of FVE on cell viability in all cell lines except Caov-3 cells, which were insensitive to FVE. This suggests that autophagy plays a major role in FVE-induced cell death. These results demonstrate that 3-MA reverses autophagy in MCF-7 and MDA-MB-231 cell lines.
During FVE-induced autophagy, LC3 is concentrated in autophagic vesicles (autophagosomes), forming punctate fluorescent structures in the cytoplasm. Overnight treatment with a low concentration of 0.1% FVE revealed distinct vesicular structures.
PI3K is involved in numerous cellular processes, including cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking. Many of these functions are linked to the ability of class I PI3 kinases to activate Akt in the PI3K/Akt/mTOR pathway. mTOR is a key effector of PI3K signaling in many human cancers, and inhibition of this pathway leads to apoptotic cell death. Furthermore, autophagy is tightly regulated by the mTOR-dependent signaling pathway, and inhibition of mTOR activity promotes autophagy.
To further explore these mechanisms, they examined the effect of FVE on the phosphorylation and activation of kinases in the PI3K-Akt-mTOR pathway in MCF-7 and MDA-MB-231 cells. FVE significantly reduced Akt phosphorylation at Ser473 and Thr308 in MCF-7 and MDA-MB-231 cells without altering total Akt levels. The effect was dose-dependent and manifested within 2 hours. A more pronounced decrease in p-Akt-Ser473 was observed after 8 hours in MDA-MB-231 cells. Consistent with Akt inactivation, FVE treatment reduced phosphorylation of the p85 regulatory subunit of PI3K in MCF-7 and MDA-MB-231 cells. They also observed that FVE-induced inhibition of Akt phosphorylation reduced mTOR complex activity, as evidenced by a significant decrease in the levels of the mTORC1 effectors p-4E-BP1 and p-p70S6K. The reduction in p-Akt-Ser/Thr by FVE was observed in T47D, OVCAR-3, HEC-1-B, RL95-2, AN3-CA, and MES-SA cell lines. However, this effect was not observed in Caov-3 cells, where p-Akt-Ser/Thr levels were undetectable. These results suggest that FVE mediates autophagic cell death by reducing the phosphorylation of key components of the PI3K/Akt/mTOR pathway.
To further examine the effect of FVE treatment on autophagy, we examined the autophagy marker LC3B. They observed a significant conversion of LC3B-I to the smaller LC3B-II, which was concentration- and time-dependent. The conversion of LC3B-I to LC3B-II by ubiquitination allows LC3B to bind to autophagic vesicles. Increased levels of phosphorylated Beclin 1 were also observed after FVE treatment. The elevated levels of phosphorylated Beclin 1 induced by FVE suggest that aberrant autophagy is a key process in promoting cell death.
In contrast to the results observed with FVE, fucoidan did not reduce p-Akt at the Ser473 or Thr308 levels. The lack of Akt inactivation and no apparent effect on proliferation suggest that fucoidan is not an active compound in FVE-mediated inhibition of PI3K/Akt/mTOR signaling in the cell lines tested.
These findings provide further evidence of the antiestrogenic activity of FVE, involving inhibition of E2-mediated ER activation and E2 synthesis. This activity suggests that FVE may confer beneficial protective effects against estrogen-dependent breast, endometrial, and ovarian cancers. Furthermore, inactivation of the PI3K/Akt/mTOR pathway reduced the viability of FVE-treated ER-positive and -negative cancer cell lines, suggesting that FVE may be useful in the treatment of some breast, endometrial, and ovarian cancers, regardless of ER status.
Source: BMC Complement Altern Med. 2016 May 28;16:151. doi: 10.1186/s12906-016-1129-6