
Fucoidan (Fucus vesiculosus), a natural sulfated polysaccharide found in various brown algae, is well known to mediate anti-cancer effects through cell cycle arrest and induction of apoptosis, but the role of the tumor suppressor p53 in the mechanism of action of fucoidan remains unclear. In this blog, I would like to introduce the research,” Induction of p53-Independent Apoptosis and G1 Cell Cycle Arrest by Fucoidan in HCT116 Human Colorectal Carcinoma Cells” by Hye Young Park et al, into the anti-cancer effects of fucoidan on two p53 isogenic HCT116 (p53+/+ and p53−/−) cell lines.
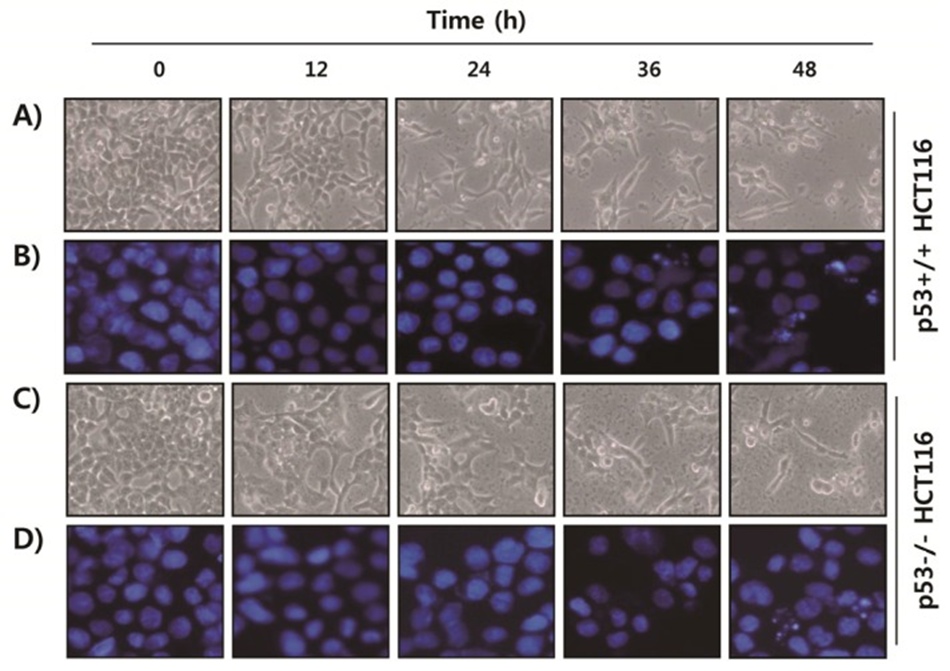
First, they evaluated the effect of fucoidan on the viability of HCT116 cells (Colorectal cancer) by MTT assay. As shown in Figure 1, fucoidan treatment gradually decreased cell viability in both p53+/+ and p53−/− HCT116 cell lines in a concentration- and time-dependent manner. Morphological observation of p53+/+ HCT116 cells showed that the cell density decreased and the number of floating dead cells increased in a time-dependent manner after fucoidan treatment (Figure 1A). DAPI staining showed that the nuclei of fucoidan-treated p53+/+ HCT116 cells were more condensed and fragmented than those of control cells, which is a phenomenon that usually occurs during apoptotic cell death (Figure 1B). These morphological changes caused by fucoidan were almost the same in p53−/− HCT116 cells (Figure 1C, D).
DNA fragmentation assays were performed, and the results showed that fragmented DNA was significantly increased after fucoidan treatment in both p53+/+ and p53−/− HCT116 cells.
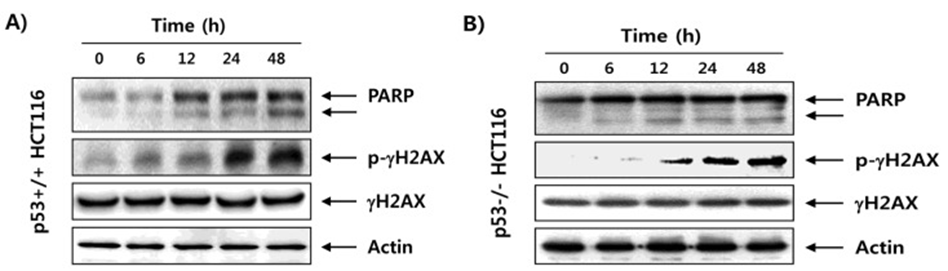
To further support the hypothesis that fucoidan induces apoptosis in HCT116 cells, we examined the effect of fucoidan on the expression of PARP. Immunoblotting results showed that the expression levels of cleaved PARP (85 kDa) were significantly increased in fucoidan-treated p53+/+ and p53−/− HCT116 cells compared with the control (Fig. 2). We also investigated the effect of fucoidan on the expression of γH2AX to examine whether the inhibition of HCT116 cell proliferation by fucoidan was associated with DNA damage induction. As shown in Fig. 2, exposure of HCT116 cells to fucoidan revealed increased phosphorylation of histone γH2AX on serine 139, a marker of DNA double-strand breaks. However, the total level of γH2AX protein was less affected by fucoidan treatment, and these effects were similar in p53+/+ and p53−/− HCT116 cells.
They next investigated whether the induction of apoptosis by fucoidan was associated with cell cycle arrest. Cell cycle phases showed normal distribution in control p53+/+ and p53−/− HCT116 cells. However, after 48 h of exposure to fucoidan, the number of cells corresponding to the G1 phase in p53+/+ HCT116 cells doubled, while the number of cells in the S and G2/M phases decreased in a time-dependent manner. Fucoidan-treated p53−/− cells showed a similar pattern to p53+/+ cells, suggesting that fucoidan-induced G1 phase cell cycle arrest is independent of p53 expression.
To clarify the mechanism of G1 cell cycle arrest caused by fucoidan, they investigated the effect of fucoidan on the expression of retinoblastoma protein (pRB) and transcription factor E2F family proteins, which function as antagonistic molecules controlling the G1/S transition. Immunoblot results showed that the expression of phosphorylated pRB in p53+/+ HCT116 cells was time-dependently decreased by fucoidan treatment without affecting the expression of E2Fs such as E2F-1 and E2F-4. We also performed co-immunoprecipitation to examine the effect of fucoidan on the interaction between pRB and E2F. The results showed that fucoidan treatment significantly enhanced the association of pRB and E2F in p53+/+ cells, suggesting that dephosphorylated pRB binds tightly to E2F.
Because the activity of G1-phase cyclin-dependent kinases (CDKs), such as CDK2, CDK4, and CDK6, is positively regulated by associated cyclins, such as D-type cyclins (cyclins D1, D2, and D3) and cyclin E, the expression of G1/S cell cycle progression-related genes in response to fucoidan treatment was next determined. The mRNA expression of cyclin D1, CDK2, and CDK4 was partially decreased in a time-dependent manner by fucoidan treatment in both p53+/+ and p53−/− HCT116 cell lines, whereas the expression levels of cyclin E and CDK6 were unchanged. Following transcriptional regulation, fucoidan downregulated the levels of cyclin D1, CDK2, and CDK4 proteins, but did not affect the expression of cyclin E and CDK6 in p53+/+ or p53−/− HCT116 cells.
In contrast to the role of CDKs and cyclins, CDKIs, such as p21WAF1/CIP1 and p27KIP1, cause cell cycle arrest by binding to cyclin/CDK complexes and inhibiting their catalytic activity. Therefore, they further investigated the effect of fucoidan on CDKI expression. The results showed that p21 and p27 mRNAs were commonly upregulated after fucoidan treatment in p53+/+ and p53−/− HCT116 cells. Meanwhile, the level of p53 tumor suppressor in p53 cells was unchanged after the same treatment, and p53 was undetectable in p53−/− HCT116 cells. Although p21 is a key transcriptional target of p53 that mediates DNA damage-induced cell cycle arrest, our results show that fucoidan treatment increases p21 expression regardless of p53 status in HCT116 cells, indicating that fucoidan regulates p21 expression in a p53-independent manner. The protein levels of p21 and p27, and their mRNA expression, were gradually increased after fucoidan treatment in both cell lines. Furthermore, co-immunoprecipitation results showed that fucoidan treatment significantly increased the binding of CDK2 and CDK4 to p21 and p27 compared to control cells. This was observed in both p53+/+ and p53−/− HCT116 cells.
The results showed that the inhibition of cell viability, induction of apoptosis, and DNA damage by fucoidan treatment were similar in the two cell lines. Flow cytometry analysis revealed that fucoidan led to a G1 arrest of cell cycle progression, which correlated with the inhibition of retinoblastoma protein (pRB) phosphorylation and the simultaneous association of pRB with transcription factors E2Fs. Furthermore, treatment with fucoidan upregulated the expression of cyclin-dependent kinase (CDK) inhibitors, such as p21WAF1/CIP1 and p27KIP1, and enhanced their binding with CDK2 and CDK4. These events occurred commonly in both cell lines, suggesting that fucoidan caused G1 arrest and apoptosis in HCT116 cells by a p53-independent mechanism. Thus, considering that most tumors show functional p53 inactivation, fucoidan may be a possible therapeutic option for cancer treatment, regardless of p53 status.
Source: Mar Drugs. 2017 May 30;15(6):154. doi: 10.3390/md15060154