
Hypertensive heart disease is a leading cause of heart failure, arrhythmia, myocardial infarction, and sudden death, making its treatment crucial. Fucoidan (FO), a natural compound extracted from seaweed, possesses both antioxidant and immunomodulatory properties. Fucoidan has also been shown to regulate apoptosis. However, it is unclear whether Fucoidan can prevent cardiac hypertrophy.
The study, “Fucoidan inhibits apoptosis and improves cardiac remodeling by inhibiting p53 transcriptional activation through USP22/Sirt 1,” by Shuai Wang et al., investigated the in vivo and in vitro effects of fucoidan on hypertrophy. I would like to bring this research to your attention.
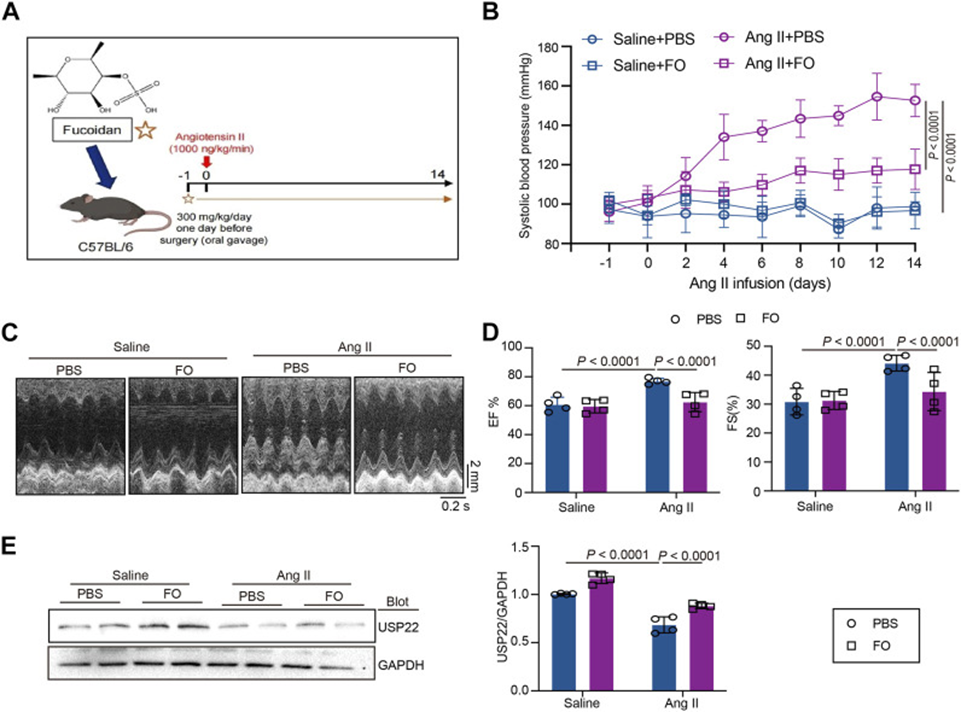
The experimental protocol involved administering 300 mg/kg of FO orally to C57BL/6 mice the day before surgery, followed by a 14-day Ang II infusion (see Figure 1A). As shown in Figure 1B, systolic blood pressure (SBP) was measured in each group, revealing that Ang II infusion significantly increased SBP, whereas Fucoidan treatment significantly reduced Ang II-induced high SBP compared with the Ang II + PBS group. Echocardiography showed that Fucoidan significantly attenuated Ang II-induced cardiac dysfunction by increasing the levels of ejection fraction (EF%) and fractional shortening (FS%) (see Figure 1C, D). Further investigation into USP22 expression revealed increased protein levels in Ang II-treated animals following Fucoidan administration (see Figure 2E). Our results indicate that Fucoidan therapy might boost USP22 expression—a protein involved in various biological processes, such as cancer—and reduce blood pressure and heart problems.
Following the initial findings, the research expanded to investigate the connection between USP22 and the cardiac hypertrophy and oxidative stress induced by Ang II. Although results showed greater left ventricular hypertrophy and higher heart weight to body weight (HW/BW) and heart weight to tibia length (HW/TL) ratios, Fucoidan therapy mitigated these findings. Furthermore, WGA staining was used to show how the cross-sectional area of cardiomyocytes changed in each group. The results showed that Fucoidan therapy reduced the size of cardiomyocytes compared to the Ang II + PBS group. In Ang II-injected animals, the expression of hypertrophy markers ANF and BNP was reduced after FO therapy. Similarly, in Ang II-injected mice, Fucoidan reduced the production of CaNA protein (a heat-resistant protein) involved in the hypertrophy signaling cascade.
Mice receiving Fucoidan treatment showed increased USP22 expression, confirming its role in the inflammatory injury and cardiac fibrosis caused by angiotensin II. The results showed that inflammatory CD68+ cells were significantly reduced in angiotensin II-injected hearts after FO treatment. The researchers found that Fucoidan significantly reduced the expression of inflammatory and fibrotic markers (IL-1β, IL-6, collagen I, and collagen III mRNA) in an angiotensin II-induced mouse model. The saline group showed lower expression of CD68 and TGF-β1 than the angiotensin II group; Fucoidan treatment in mice inhibited this angiotensin II-induced increase.
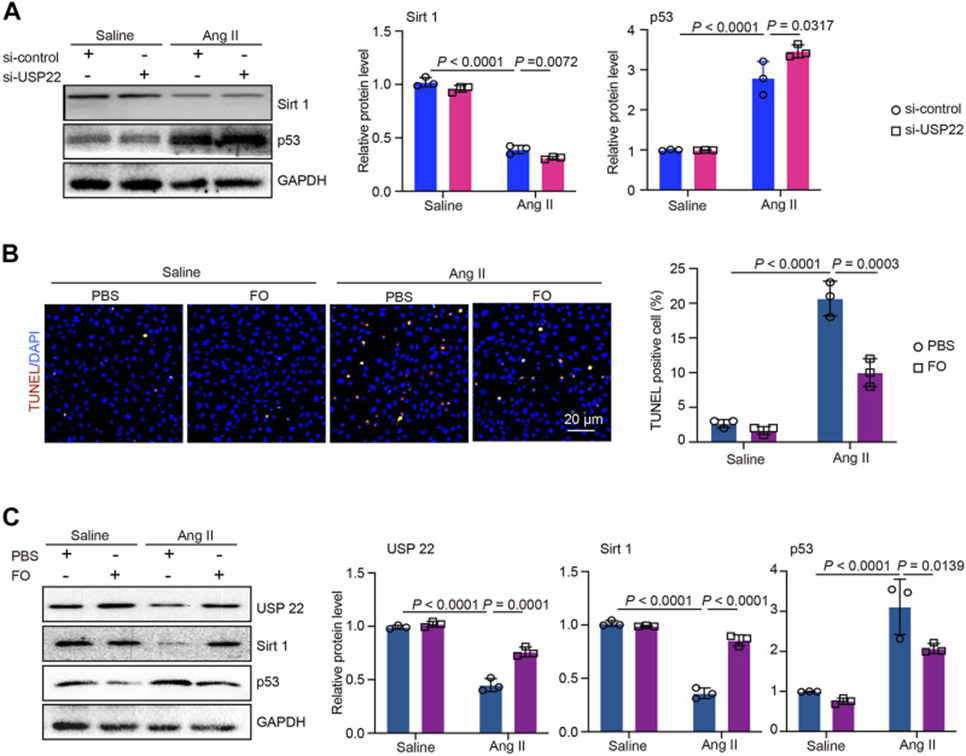
In addition, an investigation into the oxidative response in Ang II-treated animals undergoing FO therapy, utilizing DHE staining, revealed decreased expression of the oxidative markers NOX2 and NOX4. Similarly, Fucoidan reduced the production of NOX4 protein, which is involved in the oxidative signaling cascade, in Ang II-injected mice. The TUNEL assay revealed that Ang II infusion significantly increased apoptosis compared to saline controls; this effect was reduced by Fucoidan. Following this, the researchers assessed the expression of Sirt 1, p53, and Bcl-2. While saline-injected mice served as a control, Ang II-injected mice exhibited higher p53 and lower Sirt1 and Bcl-2 expression; however, FO treatment lessened these effects. These findings suggest that deubiquitination of Sirt1 by overexpression of USP22 can block p53 transcriptional activation.
The roles of USP22 and the protective effects of FO were further explored by treating AC-16 cells with si-USP22 and a si-control. Figure 2A shows that Ang II treatment caused a significant decrease in Sirt1 expression, which was further decreased by the addition of si-USP22. In contrast, p53 expression was increased by Ang II injection, and si-USP22 treatment further increased p53 expression (See Figure. 2A). Then, cells were treated with FO (60 μg/mL). TUNEL assay showed that Ang II injection increased apoptosis in AC-16 cells, and FO treatment protected this response (See Figure. 2B). Furthermore, we detected the expression of USP22, Sirt 1, and p53, and the results showed that Ang II injection decreased the expression of USP22 and Sirt 1, and increased the expression of p53, compared with the saline group (See Figure. 2C). Fucoidan treatment rescued the Ang II infusion-induced decrease in USP22 and Sirt 1 and an increase in p53 (See Figure. 2C). These results suggest that the lack of USP22 induces apoptosis in Ang II-treated cells and that Fucoidan treatment can prevent this response.
This study’s findings revealed a decreased expression of USP22 in both Ang II-injected animals and cells, suggesting a possible mechanism contributing to the development of cardiac dysfunction and remodeling. However, treatment with Fucoidan significantly increased the expression of USP22 and reduced the incidence of cardiac hypertrophy, fibrosis, inflammation, and oxidative responses. Furthermore, the administration of Fucoidan (FO) resulted in a reduction of p53 expression and apoptosis, while concurrently increasing the expression levels of Sirt 1 and Bcl-2.
By reducing Ang II-induced apoptosis through the modulation of USP22/Sirt 1 expression, FO treatment, as evidenced by this study, shows promise in improving cardiac function, establishing it as a potential targeted therapeutic strategy for patients with heart failure.
Source: Front Pharmacol. 2023 May 30;14:1164333. doi: 10.3389/fphar.2023.116433